欢迎访问《农学学报》,
农学学报 ›› 2024, Vol. 14 ›› Issue (4): 26-36.doi: 10.11923/j.issn.2095-4050.cjas2023-0092
陈昕怡( ), 刘晨艳, 华明珠, 徐欣, 冯汶祥, 汪保华, 方辉()
), 刘晨艳, 华明珠, 徐欣, 冯汶祥, 汪保华, 方辉()
收稿日期:2023-04-03
修回日期:2023-09-15
出版日期:2024-04-17
发布日期:2024-04-17
通讯作者:
作者简介:陈昕怡,女,2003年出生,江苏苏州人,本科,研究方向:玉米数量性状的遗传解析。通信地址:226019 江苏省南通市崇川区啬园路9号 南通大学主校区,E-mail:583057819@qq.com。
基金资助:
CHEN Xinyi(), LIU Chenyan, HUA Mingzhu, XU Xin, FENG Wenxiang, WANG Baohua, FANG Hui()
Received:2023-04-03
Revised:2023-09-15
Online:2024-04-17
Published:2024-04-17
摘要:
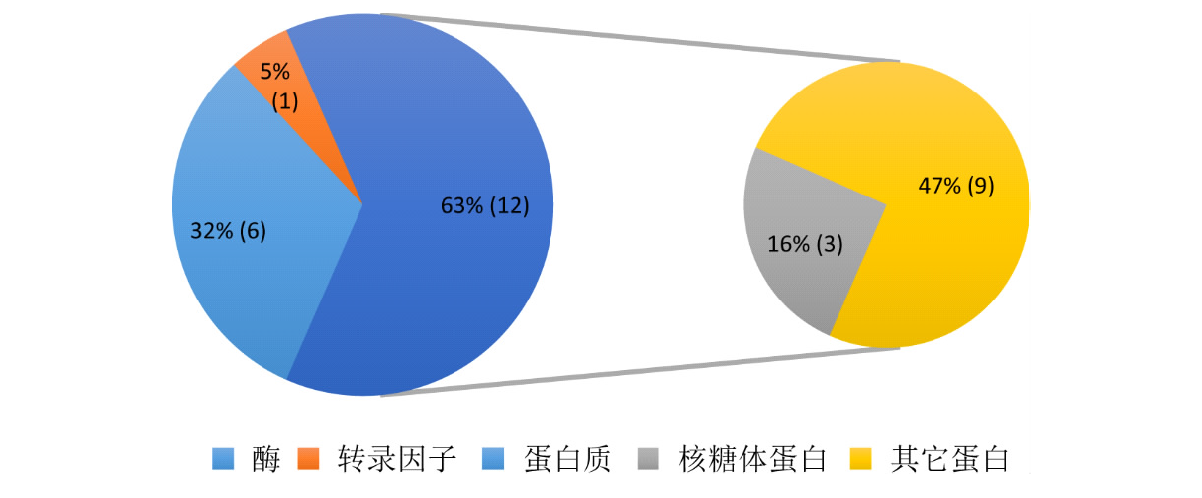
本研究旨在探索调控玉米籽粒发育的自然变异,以期为玉米产量性状的遗传改良提供科学依据。以150份遗传变异丰富的玉米自交系为材料进行研究。通过结合34342个SNP标记和3种模型,对5个籽粒相关性状进行全基因组关联分析。研究结果揭示了18个独立位点与目标性状显著关联,每个位点能够解释12.24%~15.41%的表型变异。同时,研究发现4对与籽粒长度相关的SNP之间存在显著的上位性互作,这些互作共能解释5.32%的表型变异。为了深入理解这些关联位点背后的分子机制,结合B73自交系籽粒发育的动态转录组数据和基因的功能注释,预测了19个候选基因,这些候选基因可以分为4类:6个酶、3个核糖体蛋白、1个转录因子和9个其他蛋白。这些候选基因的发现为解析玉米籽粒发育的分子机制以及改良籽粒大小和作物产量提供新的基因资源。通过本研究,我们不仅揭示了调控玉米籽粒发育的自然变异,还为玉米产量性状的遗传改良提供了新的基因资源。这些成果有望为玉米育种工作带来新的突破,提高玉米产量,从而更好地满足人类对粮食的需求。
陈昕怡, 刘晨艳, 华明珠, 徐欣, 冯汶祥, 汪保华, 方辉. 全基因组关联分析在玉米籽粒性状研究中的应用及其候选基因预测[J]. 农学学报, 2024, 14(4): 26-36.
CHEN Xinyi, LIU Chenyan, HUA Mingzhu, XU Xin, FENG Wenxiang, WANG Baohua, FANG Hui. Grain-related Traits in Maize: Genome-wide Association Analysis and Candidate Gene Prediction[J]. Journal of Agriculture, 2024, 14(4): 26-36.
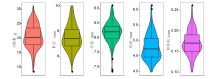
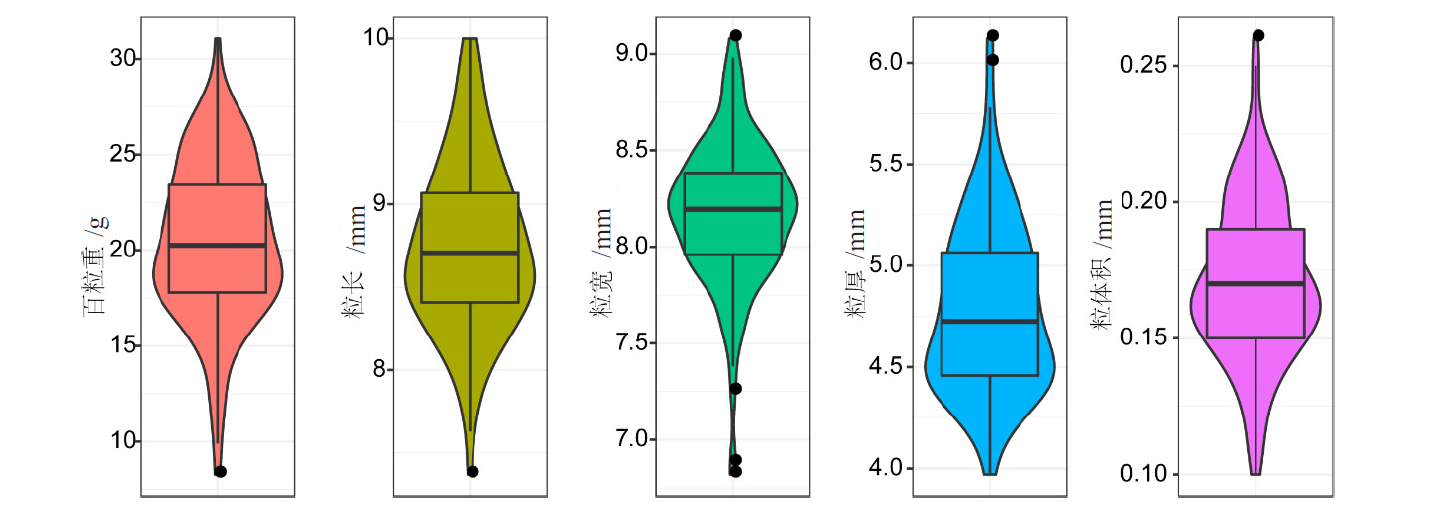
| 性状 | 自交系个数 | 最小值 | 最大值 | 平均值±标准差 | 变异系数 | 广义遗传力 | 置信区间 |
|---|---|---|---|---|---|---|---|
| HKW/g | 150 | 8.28 | 31.07 | 20.49 ± 4.12 | 20.11 | 0.995 | 0.994~0.997 |
| KL/mm | 150 | 7.37 | 10.00 | 8.75 ± 0.51 | 5.83 | 0.653 | 0.530~0.744 |
| KW/mm | 150 | 6.82 | 9.08 | 8.17 ± 0.37 | 4.53 | 0.916 | 0.886~0.938 |
| KT/mm | 150 | 3.97 | 6.12 | 4.79 ± 0.41 | 8.56 | 0.849 | 0.795~0.888 |
| KV/mL | 150 | 0.10 | 0.26 | 0.17 ± 0.03 | 17.65 | 0.523 | 0.354~0.647 |
| 性状 | 自交系个数 | 最小值 | 最大值 | 平均值±标准差 | 变异系数 | 广义遗传力 | 置信区间 |
|---|---|---|---|---|---|---|---|
| HKW/g | 150 | 8.28 | 31.07 | 20.49 ± 4.12 | 20.11 | 0.995 | 0.994~0.997 |
| KL/mm | 150 | 7.37 | 10.00 | 8.75 ± 0.51 | 5.83 | 0.653 | 0.530~0.744 |
| KW/mm | 150 | 6.82 | 9.08 | 8.17 ± 0.37 | 4.53 | 0.916 | 0.886~0.938 |
| KT/mm | 150 | 3.97 | 6.12 | 4.79 ± 0.41 | 8.56 | 0.849 | 0.795~0.888 |
| KV/mL | 150 | 0.10 | 0.26 | 0.17 ± 0.03 | 17.65 | 0.523 | 0.354~0.647 |
| HKW | KL | KW | KT | KV | |
|---|---|---|---|---|---|
| HKW | 1 | 4.62E-16 | 5.70E-16 | 3.25E-11 | 3.32E-31 |
| KL | 0.65 | 1 | 3.34E-05 | 2.05E-01 | 4.87E-12 |
| KW | 0.65 | 0.36 | 1 | 3.42E-02 | 1.03E-09 |
| KT | 0.55 | 0.12 | 0.19 | 1 | 2.23E-09 |
| KV | 0.82 | 0.57 | 0.52 | 0.51 | 1 |
| HKW | KL | KW | KT | KV | |
|---|---|---|---|---|---|
| HKW | 1 | 4.62E-16 | 5.70E-16 | 3.25E-11 | 3.32E-31 |
| KL | 0.65 | 1 | 3.34E-05 | 2.05E-01 | 4.87E-12 |
| KW | 0.65 | 0.36 | 1 | 3.42E-02 | 1.03E-09 |
| KT | 0.55 | 0.12 | 0.19 | 1 | 2.23E-09 |
| KV | 0.82 | 0.57 | 0.52 | 0.51 | 1 |
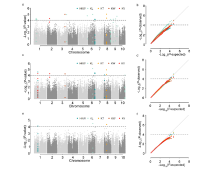
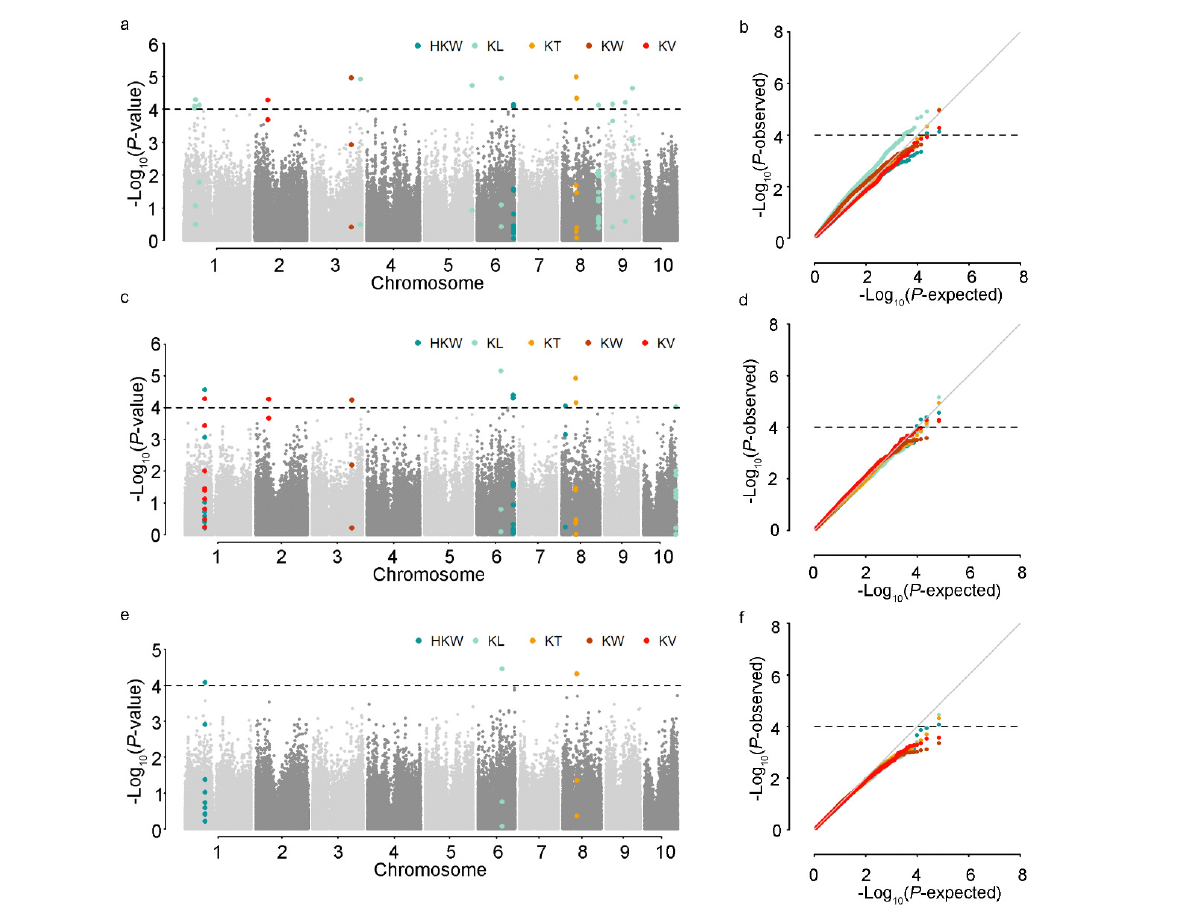
| 性状 | 标记 | 染色体 | 物理位置 | P值 | R2/% | 候选基因_V3 | 候选基因_V4 | 功能注释 | 备注 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KL | PUT-163a-4730462-2143 | 1 | 49131053 | 7.94E-05 | 12.51 | GRMZM2G038015 | Zm00001d028879 | bZIP-transcription factor 24 | ||||||||||
| KL | PZE-101070408 | 1 | 53158367 | 5.13E-05 | 13.56 | GRMZM2G107463 | Zm00001d028989 | RING/U-box superfamily protein | ||||||||||
| HKW | SYN29761 | 1 | 90219767 | 2.75E-05 | 14.29 | GRMZM2G154487 | Zm00001d029887 | Ribosomal L18p/L5e family protein | ||||||||||
| KV | PZE-102077877 | 2 | 60656421 | 5.47E-05 | 13.80 | GRMZM2G016749 | Zm00001d000124 | Protein-serine/threonine phosphatase | ||||||||||
| KW | PZE-103124207 | 3 | 181695567 | 1.10E-05 | 15.41 | GRMZM2G386991 | Zm00001d042938 | Serine/threonine-protein kinase AFC1 | ||||||||||
| KL | ZM013522-0530 | 3 | 222667473 | 1.22E-05 | 15.26 | GRMZM5G866024 | Zm00001d044376 | membrane protein | ||||||||||
| KT | SYNGENTA2236 | 4 | 5010855 | 8.07E-05 | 12.86 | GRMZM2G101502 | Zm00001d018506 | Deoxyhypusine hydroxylase | 条件 分析 | |||||||||
| KL | SYN3700 | 5 | 216163416 | 1.93E-05 | 14.62 | GRMZM2G101571 | Zm00001d018507 | bet1 sft1-related snare | ||||||||||
| KL | PZE-106060527 | 6 | 109295972 | 1.13E-05 | 15.25 | GRMZM2G048194 | Zm00001d037151 | erwinia induced protein 1 | ||||||||||
| KV | PZE-106082684 | 6 | 139912054 | 9.34E-05 | 12.71 | GRMZM2G068496 | Zm00001d037972 | 60S ribosomal protein L29 | 条件分析 | |||||||||
| GRMZM2G068323 | Zm00001d037975 | pentatricopeptide repeat-containing protein | ||||||||||||||||
| HKW | SYN38610 | 6 | 165943896 | 7.25E-05 | 12.74 | GRMZM2G132929 | Zm00001d039106 | 40S ribosomal protein S12-like | ||||||||||
| HKW | PZE-108019862 | 8 | 17889483 | 8.97E-05 | 12.47 | GRMZM2G080722 | Zm00001d008736 | Monothiol glutaredoxin-S4, mitochondrial | ||||||||||
| KT | PZE-108040469 | 8 | 65972918 | 1.05E-05 | 15.35 | GRMZM2G021331 | Zm00001d009488 | ATP synthase beta chain | ||||||||||
| KL | PZE-108113799 | 8 | 164939218 | 7.41E-05 | 14.74 | GRMZM2G049269 | Zm00001d012211 | ankyrin-like protein | ||||||||||
| KL | PZE-109030021 | 9 | 33672066 | 7.05E-05 | 12.78 | GRMZM2G065355 | Zm00001d045724 | heat shock factor-binding protein 1 | ||||||||||
| KL | SYN32340 | 9 | 90842290 | 6.11E-05 | 13.20 | GRMZM2G056166 | Zm00001d046531 | bri1-kd interacting protein 118 | ||||||||||
| KL | PZE-109077339 | 9 | 124770857 | 2.27E-05 | 14.39 | GRMZM2G113866 | Zm00001d047335 | sigma factor sigB regulation protein rsbQ | ||||||||||
| KL | PZE-110109454 | 10 | 148515533 | 9.38E-05 | 12.24 | GRMZM2G173636 | Zm00001d026639 | acyl-binding domain-containing protein 6 | ||||||||||
| 性状 | 标记 | 染色体 | 物理位置 | P值 | R2/% | 候选基因_V3 | 候选基因_V4 | 功能注释 | 备注 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KL | PUT-163a-4730462-2143 | 1 | 49131053 | 7.94E-05 | 12.51 | GRMZM2G038015 | Zm00001d028879 | bZIP-transcription factor 24 | ||||||||||
| KL | PZE-101070408 | 1 | 53158367 | 5.13E-05 | 13.56 | GRMZM2G107463 | Zm00001d028989 | RING/U-box superfamily protein | ||||||||||
| HKW | SYN29761 | 1 | 90219767 | 2.75E-05 | 14.29 | GRMZM2G154487 | Zm00001d029887 | Ribosomal L18p/L5e family protein | ||||||||||
| KV | PZE-102077877 | 2 | 60656421 | 5.47E-05 | 13.80 | GRMZM2G016749 | Zm00001d000124 | Protein-serine/threonine phosphatase | ||||||||||
| KW | PZE-103124207 | 3 | 181695567 | 1.10E-05 | 15.41 | GRMZM2G386991 | Zm00001d042938 | Serine/threonine-protein kinase AFC1 | ||||||||||
| KL | ZM013522-0530 | 3 | 222667473 | 1.22E-05 | 15.26 | GRMZM5G866024 | Zm00001d044376 | membrane protein | ||||||||||
| KT | SYNGENTA2236 | 4 | 5010855 | 8.07E-05 | 12.86 | GRMZM2G101502 | Zm00001d018506 | Deoxyhypusine hydroxylase | 条件 分析 | |||||||||
| KL | SYN3700 | 5 | 216163416 | 1.93E-05 | 14.62 | GRMZM2G101571 | Zm00001d018507 | bet1 sft1-related snare | ||||||||||
| KL | PZE-106060527 | 6 | 109295972 | 1.13E-05 | 15.25 | GRMZM2G048194 | Zm00001d037151 | erwinia induced protein 1 | ||||||||||
| KV | PZE-106082684 | 6 | 139912054 | 9.34E-05 | 12.71 | GRMZM2G068496 | Zm00001d037972 | 60S ribosomal protein L29 | 条件分析 | |||||||||
| GRMZM2G068323 | Zm00001d037975 | pentatricopeptide repeat-containing protein | ||||||||||||||||
| HKW | SYN38610 | 6 | 165943896 | 7.25E-05 | 12.74 | GRMZM2G132929 | Zm00001d039106 | 40S ribosomal protein S12-like | ||||||||||
| HKW | PZE-108019862 | 8 | 17889483 | 8.97E-05 | 12.47 | GRMZM2G080722 | Zm00001d008736 | Monothiol glutaredoxin-S4, mitochondrial | ||||||||||
| KT | PZE-108040469 | 8 | 65972918 | 1.05E-05 | 15.35 | GRMZM2G021331 | Zm00001d009488 | ATP synthase beta chain | ||||||||||
| KL | PZE-108113799 | 8 | 164939218 | 7.41E-05 | 14.74 | GRMZM2G049269 | Zm00001d012211 | ankyrin-like protein | ||||||||||
| KL | PZE-109030021 | 9 | 33672066 | 7.05E-05 | 12.78 | GRMZM2G065355 | Zm00001d045724 | heat shock factor-binding protein 1 | ||||||||||
| KL | SYN32340 | 9 | 90842290 | 6.11E-05 | 13.20 | GRMZM2G056166 | Zm00001d046531 | bri1-kd interacting protein 118 | ||||||||||
| KL | PZE-109077339 | 9 | 124770857 | 2.27E-05 | 14.39 | GRMZM2G113866 | Zm00001d047335 | sigma factor sigB regulation protein rsbQ | ||||||||||
| KL | PZE-110109454 | 10 | 148515533 | 9.38E-05 | 12.24 | GRMZM2G173636 | Zm00001d026639 | acyl-binding domain-containing protein 6 | ||||||||||
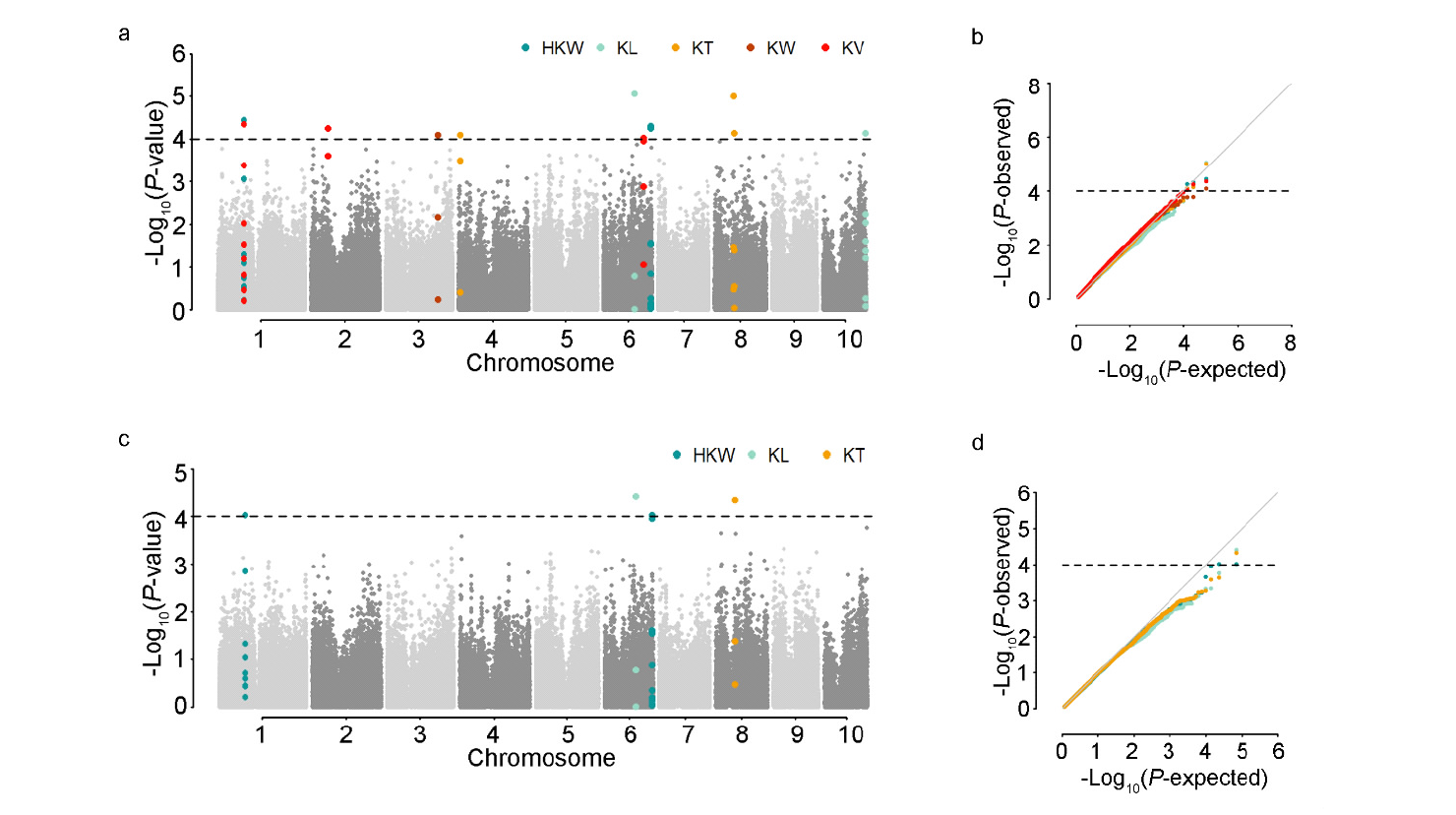
| 性状 | SNP1 | SNP2 | P值 | add_R2/%a | epi_R2/%b |
|---|---|---|---|---|---|
| KL | PUT-163a-4730462-2143 | ZM013522-0530 | 0.02 | 53.39 | 5.32 |
| KL | PUT-163a-4730462-2143 | PZE-109077339 | 0.03 | 53.39 | 5.32 |
| KL | PZE-101070408 | PZE-110109454 | 0.02 | 53.39 | 5.32 |
| KL | PZE-108113799 | PZE-109077339 | 0.01 | 53.39 | 5.32 |
| KV | - | - | - | 20.13 | - |
| KT | - | - | - | 22.63 | - |
| HKW | - | - | - | 22.79 | - |
| KV | - | - | - | 15.41 | - |
| 性状 | SNP1 | SNP2 | P值 | add_R2/%a | epi_R2/%b |
|---|---|---|---|---|---|
| KL | PUT-163a-4730462-2143 | ZM013522-0530 | 0.02 | 53.39 | 5.32 |
| KL | PUT-163a-4730462-2143 | PZE-109077339 | 0.03 | 53.39 | 5.32 |
| KL | PZE-101070408 | PZE-110109454 | 0.02 | 53.39 | 5.32 |
| KL | PZE-108113799 | PZE-109077339 | 0.01 | 53.39 | 5.32 |
| KV | - | - | - | 20.13 | - |
| KT | - | - | - | 22.63 | - |
| HKW | - | - | - | 22.79 | - |
| KV | - | - | - | 15.41 | - |
| [1] |
doi: 10.1371/journal.pone.0066428 URL |
| [2] |
doi: 10.1104/pp.19.00546 URL |
| [3] |
doi: 10.1111/jipb.v62.6 URL |
| [4] |
doi: 10.3389/fpls.2020.00814 URL |
| [5] |
doi: 10.1104/pp.92.4.881 URL |
| [6] |
doi: 10.1073/pnas.1904995116 URL |
| [7] |
doi: 10.1007/s11103-016-0543-y pmid: 27709320 |
| [8] |
doi: 10.1093/mp/ssq057 pmid: 20924026 |
| [9] |
doi: 10.1104/pp.109.150193 pmid: 20044449 |
| [10] |
doi: 10.1534/genetics.116.196105 pmid: 27815362 |
| [11] |
doi: 10.1534/genetics.116.199331 URL |
| [12] |
doi: 10.1016/j.molp.2016.08.008 URL |
| [13] |
doi: 10.1016/j.molp.2016.10.016 URL |
| [14] |
doi: 10.1105/tpc.18.00266 pmid: 30201823 |
| [15] |
doi: 10.1105/tpc.006726 URL |
| [16] |
doi: 10.1371/journal.pone.0153428 URL |
| [17] |
龚玉林, 贺丹, 卫芸芸, 等. 真菌遗传学方法研究进展[J]. 菌物研究, 2019, 17(3):173-179.
|
| [18] |
doi: 10.1371/journal.pone.0089645 URL |
| [19] |
doi: 10.1007/s00438-015-1121-8 URL |
| [20] |
doi: 10.1186/1471-2229-13-1 URL |
| [21] |
doi: 10.1007/s10681-013-0901-7 URL |
| [22] |
doi: 10.1007/s00122-016-2717-z URL |
| [23] |
doi: 10.1038/s41588-019-0427-6 pmid: 31152161 |
| [24] |
doi: 10.1007/s00122-011-1532-9 pmid: 21286680 |
| [25] |
doi: 10.1371/journal.pone.0124779 URL |
| [26] |
doi: S0168-9452(15)30079-0 pmid: 26566847 |
| [27] |
doi: 10.1007/s11032-015-0425-z URL |
| [28] |
doi: 10.1186/s12870-015-0700-5 URL |
| [29] |
doi: 10.1104/pp.17.00708 pmid: 28811335 |
| [30] |
doi: 10.3389/fpls.2017.02190 pmid: 29312420 |
| [31] |
doi: 10.1104/pp.16.01516 pmid: 28153923 |
| [32] |
doi: 10.1111/pbi.13645 pmid: 34077617 |
| [33] |
doi: 10.1126/science.abg7985 URL |
| [34] |
doi: 10.1126/science.1177837 pmid: 19965431 |
| [35] |
doi: 10.1038/ng.2313 |
| [36] |
|
| [37] |
doi: 10.1371/journal.pgen.1004573 URL |
| [38] |
doi: 10.1111/pbi.12821 pmid: 28796926 |
| [39] |
doi: 10.1111/pbi.13188 pmid: 31199064 |
| [40] |
doi: 10.1007/s00438-019-01631-2 pmid: 31807910 |
| [41] |
doi: 10.1007/s00122-017-2867-7 pmid: 28215025 |
| [42] |
代力强, 吴律, 董青松, 等. 玉米籽粒长度的全基因组关联分析[J]. 西北农林科技大学学报(自然科学版), 2018, 6.
|
| [43] |
渠建洲, 冯文豪, 张兴华, 等. 基于全基因组关联分析解析玉米籽粒大小的遗传结构[J]. 作物学报, 2022, 48(2):304-319.
doi: 10.3724/SP.J.1006.2022.13002 |
| [44] |
肖颖妮, 李高科, 李坤, 等. 甜玉米籽粒体积和粒重的全基因组关联分析[J]. 中国农业大学学报, 2022, 27(7):12-25.
|
| [45] |
doi: 10.1186/s12870-020-02777-7 |
| [46] |
doi: 10.2135/cropsci1985.0011183X002500010046x URL |
| [47] |
doi: 10.1111/tpj.14539 pmid: 31529523 |
| [48] |
doi: 10.1086/519795 URL |
| [49] |
doi: 10.1371/journal.pone.0028334 URL |
| [50] |
单婷玉, 施雯, 王翌婷, 等. 玉米盐胁迫相关性状全基因组关联分析及候选基因预测[J]. 遗传, 2021, 43(12):1159-1169.
|
| [51] |
doi: 10.1186/1471-2105-12-1 |
| [52] |
doi: 10.1093/bioinformatics/btm308 pmid: 17586829 |
| [53] |
doi: 10.1038/ng1702 pmid: 16380716 |
| [54] |
doi: 10.1093/bioinformatics/10.2.189 URL |
| [55] |
|
| [56] |
doi: 10.1104/pp.114.240689 pmid: 25037214 |
| [57] |
doi: 10.1007/s00726-007-0525-0 pmid: 17476569 |
| [58] |
doi: S1674-2052(16)30308-2 pmid: 28039028 |
| [59] |
doi: 10.1038/ng.3636 |
| [60] |
doi: 10.1038/ncomms9326 |
| [61] |
doi: 10.1016/j.molp.2021.11.007 URL |
| [62] |
doi: 10.1038/s41477-019-0565-y |
| [63] |
doi: 10.1111/pbi.13607 pmid: 33934485 |
| [64] |
doi: 10.1038/s41467-019-14027-y |
| [65] |
doi: 10.1038/s41467-021-25001-y pmid: 34354054 |
| [66] |
doi: 10.1038/s41588-019-0503-y |
| [67] |
doi: 10.1073/pnas.1310949110 URL |
| [68] |
丁安明, 屈旭, 李凌, 等. 植物PPR蛋白家族研究进展[J]. 中国农学通报, 2014, 30(9):218-224.
|
| [69] |
doi: 10.1146/annurev-arplant-050213-040159 pmid: 24471833 |
| [70] |
doi: 10.1105/tpc.112.106781 URL |
| [71] |
doi: 10.1111/nph.2017.214.issue-2 URL |
| [72] |
doi: 10.1111/tpj.2017.91.issue-1 URL |
| [73] |
doi: 10.1534/genetics.117.300602 URL |
| [74] |
doi: 10.1105/tpc.112.099051 URL |
| [75] |
doi: 10.1016/j.molp.2017.09.009 URL |
| [76] |
|
| [77] |
doi: 10.1080/15476286.2020.1817267 URL |
| [78] |
doi: 10.1046/j.1365-313X.2003.01942.x URL |
| [79] |
doi: 10.1104/pp.103.030767 URL |
| [80] |
doi: 10.1104/pp.15.01722 pmid: 26645456 |
| [81] |
doi: 10.1111/nph.15057 pmid: 29479724 |
| [1] | 刘蔚霞, 路笃旭, 卢振宇, 张超, 翟吉庆, 翟乃家, 王光明. 淄博地区适宜与玉米间作种植的大豆品种初报[J]. 农学学报, 2024, 14(4): 7-13. |
| [2] | 边金霞, 王平. 旱作区化肥减量配施有机肥对粮饲兼用玉米生长及产量的影响[J]. 农学学报, 2024, 14(3): 13-20. |
| [3] | 李艳兰, 普光发, 张娜, 徐学忠, 胡新洲, 柴梦婷, 张雪松, 刘坚坚, 李祥, 陈梦丽, 张斌, 任晶梅, 杨进成. 稀植鲜食蚕豆品种产量与农艺性状相关性分析[J]. 农学学报, 2024, 14(3): 28-33. |
| [4] | 张国, 于居龙, 凌鸿, 姚克兵, 余向阳, 程金金, 朱凤, 周晨, 束兆林, 赵来成. 二氢卟吩铁浸种对水稻生长与增产效应研究[J]. 农学学报, 2024, 14(3): 34-39. |
| [5] | 高润, 吴波, 陈燕, 刘逸, 张进红, 许瑞轩, 王国良. 黄淮海地区苜蓿田夏季套作青贮玉米研究初探[J]. 农学学报, 2024, 14(2): 1-6. |
| [6] | 高鹏, 余正军, 杨小敏, 王涛, 李丹妮. 汉中市土壤有效锌的含量分布及影响因素分析[J]. 农学学报, 2024, 14(2): 36-41. |
| [7] | 颜为, 李晓靖, 姜玉玮, 黄萌, 刘波, 张春艳, 崔振岭, 薛艳芳. 播期和叶面肥喷施对不同玉米品种产量和籽粒矿质元素含量的影响[J]. 农学学报, 2024, 14(1): 1-9. |
| [8] | 杨美丽, 王帮太, 鹿红卫, 苏玉杰, 赵树政, 程建梅, 王静, 郭华, 王志红, 秦贵文. 玉米自交系花丝及其结实性动态研究[J]. 农学学报, 2024, 14(1): 10-14. |
| [9] | 吴豪, 文羿, 林云红, 熊茜, 徐炜, 杨茂松, 周有海, 王戈, 杜宇, 白羽祥, 王娜, 周鹏. 密度和留叶数对烤烟‘NC297’生长及产质量的影响[J]. 农学学报, 2024, 14(1): 15-21. |
| [10] | 赵凯琴, 张玉松, 燕林祥, 李根泽, 李庆刚, 罗延青. 种植密度和施氮量对油菜生长发育及产量的影响[J]. 农学学报, 2024, 14(1): 22-28. |
| [11] | 赵明明, 胡新燕, 李卫华, 李可, 王康, 陈晓光. 早熟优质宜机采棉花新品种‘徐棉608’的选育[J]. 农学学报, 2024, 14(1): 29-33. |
| [12] | 胡杨, 段斌, 方玲, 何世界, 李慧龙, 宋晓华, 余新春, 常幸远. 苗期不同浓度多效唑处理对豫南粳稻秧苗素质及产量构建的影响[J]. 农学学报, 2023, 13(9): 1-7. |
| [13] | 黄慧. 有机无机肥配施对夏玉米根系分布及产量形成的调控[J]. 农学学报, 2023, 13(9): 13-17. |
| [14] | 周恩强, 姚梦楠, 周瑶, 薛冬, 王永强, 赵娜, 魏利斌, 王学军, 缪亚梅. 不同播期对鲜食春大豆品种产量和农艺性状的影响[J]. 农学学报, 2023, 13(9): 25-30. |
| [15] | 杨宁, 赵士花, 种冬冬, 李静欣, 王秀芹, 董玲霞, 杜宗清, 李瑞. 不同品种不同播期对小麦黄花叶病毒病的抗性及产量的影响[J]. 农学学报, 2023, 13(9): 8-12. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||